
Din cuprinsul articolului
Sindromul Hutchinson-Gilford, cunoscut și sub numele de progerie, este o afecțiune genetică rară care provoacă îmbătrânirea prematură la copii. Această boală afectează aproximativ 1 din 4-8 milioane de nou-născuți.
Sindromul Hutchinson-Gilford este o afecțiune genetică rară și devastatoare, caracterizată prin îmbătrânirea prematură a copiilor.
Înțelegerea cauzelor și simptomelor acestui sindrom este esențială pentru diagnosticarea și gestionarea adecvată a bolii. Deși nu există un tratament curativ, monitorizarea atentă și tratamentele simptomatice pot îmbunătăți calitatea vieții pacienților afectați.
Sindromul Hutchinson-Gilford. Cauzele apariției
Sindromul Hutchinson-Gilford este cauzat de o mutație în gena LMNA, care codifică proteina lamin A. Această proteină este esențială pentru structura și funcționarea normală a nucleului celular.
Mutația duce la producerea unei forme anormale a proteinei, numită progerină, care afectează stabilitatea nucleului și determină îmbătrânirea prematură a celulelor.
Această mutație apare de obicei spontan și nu este moștenită de la părinți. Progerina acumulată în celule perturbă funcționarea normală a acestora, ducând la manifestările clinice ale sindromului.
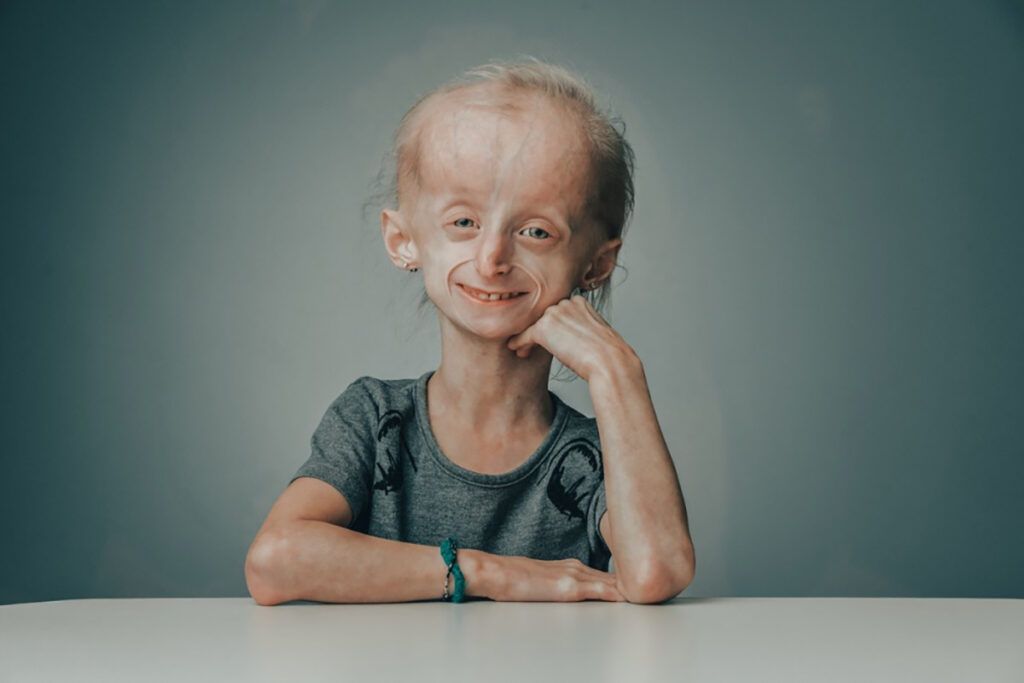
Simptomele Sindromului Hutchinson-Gilford
Simptomele sindromului Hutchinson-Gilford devin evidente în primele luni sau ani de viață și includ:
- Îmbătrânire prematură: Copiii afectați dezvoltă semne de îmbătrânire rapidă, cum ar fi pielea subțire și fragilă, pierderea părului (alopecie) și apariția ridurilor, potrivit doc.ro.
- Creștere lentă: Acești copii cresc mai încet decât media și au o statură mică.
- Aspect facial caracteristic: Ochii proeminenți, nasul subțire și îngust, buzele subțiri și bărbia mică sunt trăsături comune.
- Probleme cardiace: Ateroscleroza (îngroșarea și întărirea arterelor) este frecventă, crescând riscul de atac de cord sau accident vascular cerebral la o vârstă fragedă.
- Probleme scheletice: Fragilitatea oaselor și rigiditatea articulațiilor sunt simptome comune.
- Pierderea grăsimii subcutanate: Copiii cu acest sindrom au un aspect îmbătrânit din cauza pierderii grăsimii sub piele.
Sindromul Hutchinson-Gilford nu afectează dezvoltarea intelectuală sau abilitățile motorii, cum ar fi mersul pe jos. Cu toate acestea, complicațiile severe, cum ar fi problemele cardiace, pot reduce semnificativ speranța de viață, care este în medie de aproximativ 14,5 ani.
Cum se poate diagnostica sindromul Hutchinson-Gilford
Diagnosticul sindromului Hutchinson-Gilford (progerie) se bazează pe o combinație de evaluări clinice și teste genetice. Iată pașii principali pentru diagnosticare:
1. Evaluarea clinică: Medicul va examina copilul pentru a identifica semnele caracteristice ale sindromului, cum ar fi îmbătrânirea prematură, creșterea lentă, aspectul facial distinctiv și alte simptome fizice.
2. Istoricul medical: Se va colecta un istoric medical detaliat pentru a înțelege evoluția simptomelor și pentru a exclude alte afecțiuni care ar putea prezenta simptome similare.
3. Teste genetice: Diagnosticul definitiv se face prin teste genetice care identifică mutația specifică în gena LMNA. Aceste teste pot include:
- Secvențierea ADN-ului: Aceasta implică analiza ADN-ului copilului pentru a detecta mutația în gena LMNA.
- Testele de amplificare a fragmentelor: Acestea pot ajuta la identificarea mutațiilor specifice prin amplificarea și analizarea unor segmente de ADN.
4. Teste suplimentare: În unele cazuri, medicul poate recomanda teste suplimentare pentru a evalua starea generală de sănătate a copilului și pentru a monitoriza complicațiile asociate, cum ar fi problemele cardiace și scheletice.
Diagnosticul precoce este esențial pentru gestionarea eficientă a sindromului Hutchinson-Gilford și pentru îmbunătățirea calității vieții pacienților afectați.

Există vreun tratament pentru această afecțiune
În prezent, nu există un tratament curativ pentru sindromul Hutchinson-Gilford (progerie), dar există opțiuni de tratament care pot ajuta la gestionarea simptomelor și la îmbunătățirea calității vieții pacienților. Iată câteva dintre acestea:
1. Medicamente:
- Lonafarnib: Acesta este un inhibitor de farnesiltransferază aprobat de FDA pentru tratamentul progeriei. Lonafarnib ajută la reducerea acumulării de progerină în celule, ceea ce poate încetini progresia bolii și poate prelungi speranța de viață.
- Statine și inhibitori de enzime de conversie a angiotensinei (ECA): Aceste medicamente pot fi utilizate pentru a gestiona problemele cardiovasculare asociate cu progeria.
2. Terapie fizică și ocupațională: Aceste terapii pot ajuta la menținerea mobilității și la îmbunătățirea funcției motorii, reducând rigiditatea articulațiilor și îmbunătățind calitatea vieții.
3. Monitorizarea regulată: Evaluările medicale periodice sunt esențiale pentru a monitoriza starea de sănătate a pacienților și pentru a detecta și trata prompt complicațiile, cum ar fi problemele cardiace și scheletice.
4. Intervenții chirurgicale: În unele cazuri, pot fi necesare intervenții chirurgicale pentru a corecta problemele ortopedice sau pentru a trata complicațiile cardiovasculare.
5. Suport psihologic: Consilierea și suportul psihologic sunt importante pentru pacienți și familiile lor, ajutându-i să facă față provocărilor emoționale și psihologice asociate cu această afecțiune rară.
Deși tratamentele disponibile nu pot vindeca progeria, ele pot contribui semnificativ la îmbunătățirea calității vieții și la prelungirea speranței de viață a pacienților.
Care sunt complicațiile asociate cu sindromul Hutchinson-Gilford
Sindromul Hutchinson-Gilford (progerie) este asociat cu o serie de complicații severe, care afectează în mod semnificativ calitatea vieții și speranța de viață a pacienților. Iată câteva dintre cele mai comune complicații:
1. Probleme cardiovasculare:
- Ateroscleroza: Îngroșarea și întărirea arterelor, care poate duce la boli coronariene.
- Hipertensiune arterială: Tensiunea arterială ridicată este frecventă și poate agrava problemele cardiace.
- Insuficiență cardiacă: Deteriorarea inimii poate duce la insuficiență cardiacă, care este o cauză principală de deces la pacienții cu progerie.
2. Accident vascular cerebral: Riscul de accident vascular cerebral este crescut din cauza problemelor vasculare.
3. Probleme scheletice:
- Osteoporoză: Fragilitatea oaselor, care crește riscul de fracturi.
- Rigiditatea articulațiilor: Articulațiile devin rigide și dureroase, limitând mobilitatea.
4. Probleme dermatologice:
- Piele subțire și fragilă: Pielea devine foarte subțire și predispusă la răni și infecții.
- Pierderea părului: Alopecia este comună, iar pacienții își pierd părul de pe scalp, sprâncene și gene.
5. Probleme de creștere: Statură mică: Copiii cu progerie au o creștere lentă și rămân mult mai mici decât colegii lor de vârstă.
6. Probleme dentare:
- Întârzierea erupției dentare: Dinții pot erupe mai târziu decât în mod normal.
- Probleme dentare: Dinții pot fi deformați sau lipsiți, ceea ce necesită îngrijire dentară specializată.
7. Complicații metabolice: Rezistența la insulină: Pacienții pot dezvolta rezistență la insulină, ceea ce poate duce la diabet.
Monitorizarea regulată și tratamentele simptomatice sunt esențiale pentru a gestiona aceste complicații și pentru a îmbunătăți calitatea vieții pacienților cu sindromul Hutchinson-Gilford.






